|
GLYLIB
0.3.0b
|
|
GLYLIB
0.3.0b
|
#include <molecules.h>
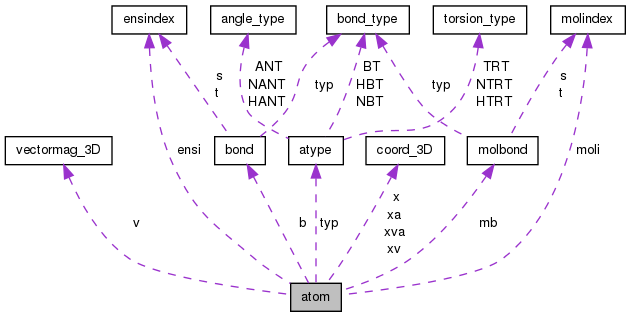
Public Attributes | |
| int | n |
| atom number or other identifying index | |
| char * | N |
| atom name | |
| char * | T |
| atom type name | |
| char * | D |
| free-form description | |
| char * | E |
| atom element | |
| double | m |
| atom mass -- units defined per program needs | |
| int | t |
| type number -- must correspond to assignments of "atype" (see) | |
| atype * | typ |
| pointer to atype structure | |
| molindex | moli |
| the molecule index for this atom (address) | |
| int | nb |
| number of bonds within this residue (deprecated, soon to go away) | |
| bond * | b |
| bond structures (nb of these) | |
| int | nmb |
| number of bonds to other residues or molecules | |
| molbond * | mb |
| nmb of these (pointers to the structures) | |
| int | mTi |
| Index into the molecule's connection tree for this atom (m.aT) | |
| int | rTi |
| Index into the residue's connection tree for this atom (r.aT) | |
| coord_3D | x |
| atom's main coordinates | |
| coord_3D | xv |
| atom's main velocity | |
| int | nalt |
| number of alternate coordinate sets | |
| coord_3D * | xa |
| nalt of alternate coords | |
| int | nxva |
| number of alternate velocities defined | |
| coord_3D * | xva |
| nxva alternate velocities | |
| int | nvec |
| number of vector sets | |
| vectormag_3D * | v |
| vector sets | |
| int | nch |
| number of charges | |
| double * | ch |
| nch charges for this atom | |
| int | ni |
| number of other indices | |
| int * | i |
| other indices, as needed (ni of these) | |
| int | nd |
| number of double-precision parameters | |
| double * | d |
| other parameters, as needed (nd of these) | |
| int | nensi |
| number of ensemble indices | |
| ensindex * | ensi |
| list of ensemble indices | |
| char * | sres |
| name of some other/original residue to which this atom belongs | |
| int | nOD |
| number of other descriptors | |
| char ** | OD |
| the nOD descriptors | |
| int | nVP |
| number of void structures | |
| char * | cID |
| chain identifier | |
| void * | VP |
| nVP of these, whatever they may be | |
Definition at line 139 of file molecules.h.
bond structures (nb of these)
Definition at line 150 of file molecules.h.
Referenced by initialize_atom().
| double* atom::ch |
nch charges for this atom
Definition at line 164 of file molecules.h.
Referenced by get_molecule_point_charge_dipole(), initialize_atom(), and parse_amber_prmtop().
| char* atom::cID |
| char* atom::D |
| double* atom::d |
other parameters, as needed (nd of these)
Definition at line 168 of file molecules.h.
Referenced by initialize_atom().
| char* atom::E |
list of ensemble indices
Definition at line 170 of file molecules.h.
Referenced by initialize_atom(), and parse_amber_prmtop().
| int* atom::i |
other indices, as needed (ni of these)
Definition at line 166 of file molecules.h.
Referenced by dprint_atom(), dXprint_atom(), get_assembly_PDB_ATOM_lines(), get_ensemble_PDB_ATOM_lines(), get_PDB_line_for_ATOM(), and initialize_atom().
| double atom::m |
atom mass -- units defined per program needs
Definition at line 145 of file molecules.h.
Referenced by follow_molecule_atom_nodes_from_bonds(), get_assembly_molecule_COM(), get_ensemble_COM(), get_molecule_COM(), get_residue_COM(), getAssembly(), initialize_atom(), and parse_amber_prmtop().
nmb of these (pointers to the structures)
Definition at line 152 of file molecules.h.
Referenced by follow_molecule_atom_molbonds_for_contree(), follow_molecule_atom_nodes_from_bonds(), follow_residue_atom_nodes_from_bonds(), get_molecule_PDB_ATOM_lines(), initialize_atom(), parse_amber_prmtop(), set_assembly_atom_molbonds_from_PDB_CONECT(), and set_molecule_residue_molbonds().
the molecule index for this atom (address)
Definition at line 148 of file molecules.h.
Referenced by add_assembly_to_ensemble(), find_assembly_top_level_atoms_by_n(), find_residue_atoms_by_N(), follow_molecule_atom_molbonds_for_contree(), parse_amber_prmtop(), set_assembly_atom_molbonds_from_PDB_CONECT(), set_connection_tree_molecule_atoms(), and set_residue_molindexes().
| int atom::mTi |
Index into the molecule's connection tree for this atom (m.aT)
Definition at line 153 of file molecules.h.
Referenced by follow_molecule_atom_molbonds_for_contree(), follow_molecule_atom_nodes_from_bonds(), initialize_atom(), set_connection_tree_molecule_atoms(), and set_molecule_atom_nodes_from_bonds().
| int atom::n |
atom number or other identifying index
Definition at line 140 of file molecules.h.
Referenced by get_PDB_line_for_ATOM(), get_residue_PDB_ATOM_lines(), initialize_atom(), load_dlg_mol(), outputAsmblPDB(), outputMolPDB(), parse_amber_prmtop(), read_prepatom(), and set_assembly_atom_molbonds_from_PDB_CONECT().
| char* atom::N |
atom name
Definition at line 141 of file molecules.h.
Referenced by initialize_atom(), outputAsmblPDB(), outputMolPDB(), parse_amber_prmtop(), and read_prepatom().
| int atom::nalt |
number of alternate coordinate sets
Definition at line 157 of file molecules.h.
Referenced by add_trajcrds_to_prmtop_assembly(), get_assembly_PDB_ATOM_lines(), get_ensemble_PDB_ATOM_lines(), initialize_atom(), and load_dlg_mol().
| int atom::nb |
number of bonds within this residue (deprecated, soon to go away)
Definition at line 149 of file molecules.h.
Referenced by deallocateAtom(), and initialize_atom().
| int atom::nch |
number of charges
Definition at line 163 of file molecules.h.
Referenced by initialize_atom(), and parse_amber_prmtop().
| int atom::nd |
number of double-precision parameters
Definition at line 167 of file molecules.h.
Referenced by initialize_atom().
| int atom::nensi |
number of ensemble indices
Definition at line 169 of file molecules.h.
Referenced by initialize_atom(), and parse_amber_prmtop().
| int atom::ni |
number of other indices
Definition at line 165 of file molecules.h.
Referenced by get_assembly_PDB_ATOM_lines(), get_ensemble_PDB_ATOM_lines(), getAssembly(), and initialize_atom().
| int atom::nmb |
number of bonds to other residues or molecules
Definition at line 151 of file molecules.h.
Referenced by deallocateAtom(), follow_molecule_atom_molbonds_for_contree(), follow_molecule_atom_nodes_from_bonds(), follow_residue_atom_nodes_from_bonds(), get_molecule_PDB_ATOM_lines(), initialize_atom(), parse_amber_prmtop(), set_assembly_atom_molbonds_from_PDB_CONECT(), and set_molecule_residue_molbonds().
| int atom::nOD |
number of other descriptors
Definition at line 172 of file molecules.h.
Referenced by deallocateAtom(), and initialize_atom().
| int atom::nvec |
number of vector sets
Definition at line 161 of file molecules.h.
Referenced by add_trajcrds_to_prmtop_assembly(), and initialize_atom().
| int atom::nVP |
number of void structures
Definition at line 174 of file molecules.h.
Referenced by initialize_atom(), and read_prepatom().
| int atom::nxva |
number of alternate velocities defined
Definition at line 159 of file molecules.h.
Referenced by initialize_atom().
| char** atom::OD |
| int atom::rTi |
Index into the residue's connection tree for this atom (r.aT)
Definition at line 154 of file molecules.h.
Referenced by follow_residue_atom_nodes_from_bonds(), initialize_atom(), and set_residue_atom_nodes_from_bonds().
| char* atom::sres |
name of some other/original residue to which this atom belongs
Definition at line 171 of file molecules.h.
| char* atom::T |
atom type name
Definition at line 142 of file molecules.h.
Referenced by initialize_atom(), parse_amber_prmtop(), and read_prepatom().
| int atom::t |
type number -- must correspond to assignments of "atype" (see)
Definition at line 146 of file molecules.h.
Referenced by get_assembly_molecule_COM(), get_ensemble_COM(), get_molecule_COM(), get_residue_COM(), initialize_atom(), load_dlg_mol(), parse_amber_prmtop(), read_prepatom(), and read_prepres().
pointer to atype structure
Definition at line 147 of file molecules.h.
Referenced by deallocateAtom(), and initialize_atom().
vector sets
Definition at line 162 of file molecules.h.
Referenced by add_trajcrds_to_prmtop_assembly(), initialize_atom(), normalize_molecule_vectors(), and rotate_vector_to_Z_M().
| void* atom::VP |
nVP of these, whatever they may be
Definition at line 176 of file molecules.h.
Referenced by read_prepatom().
atom's main coordinates
Definition at line 155 of file molecules.h.
Referenced by dprint_atom(), dXprint_atom(), get_assembly_molecule_COM(), get_ensemble_COM(), get_molecule_COM(), get_molecule_point_charge_dipole(), get_residue_COM(), initialize_atom(), load_dlg_mol(), outputAsmblPDB(), outputMolPDB(), rollAssembly(), rollMolecule(), rotate_vector_to_Z_M(), shift_molecule_atoms_by_vector_scale(), translate_ensemble_by_XYZ(), translate_molecule_by_XYZ(), translate_residue_by_XYZ(), and translate_zero_to_coord_M().
nalt of alternate coords
Definition at line 158 of file molecules.h.
Referenced by add_trajcrds_to_prmtop_assembly(), get_assembly_molecule_COM(), get_ensemble_COM(), get_molecule_COM(), get_molecule_point_charge_dipole(), get_residue_COM(), initialize_atom(), load_dlg_mol(), rotate_vector_to_Z_M(), shift_molecule_atoms_by_vector_scale(), translate_ensemble_by_XYZ(), translate_molecule_by_XYZ(), translate_residue_by_XYZ(), and translate_zero_to_coord_M().
nxva alternate velocities
Definition at line 160 of file molecules.h.
Referenced by initialize_atom().
 1.7.6.1
1.7.6.1 
